
INTRODUÇÃO
Há um grande número de exames laboratoriais disponíveis comercialmente que têm utilidade na avaliação do paciente com suspeita de doença hepática ou na investigação da sua causa. Os exames podem ser classificados de modo didático em:
- Testes para avaliação de lesão hepatocelular (destruição de hepatócitos);
- Testes para avaliação do fluxo biliar e lesão de vias biliares;
- Testes para avaliação da função de síntese do fígado;
- Testes para avaliação de complicações e estágio da cirrose;
- Testes para investigação da etiologia (causa) da doença hepática (abordados em outros textos).
O termo “função hepática” geralmente é utilizado erroneamente na prática clínica para descrever um conjunto de exames laboratoriais que não investigam apenas a função do fígado, mas também a presença de lesão hepatocelular e de vias biliares. Costuma incluir AST, ALT, fosfatase alcalina, GGT, albumina, bilirrubinas total e frações e atividade da protrombina.

Foto por Pixabay em Pexels.com
- Testes para avaliação de lesão hepatocelular
1.1 Aminotransferases
1.1.1 Aspartato aminotransferase (AST)
- também pode ser chamada de transaminase glutâmico oxaloacética (TGO)
- é uma enzima que catalisa a reação: aspartato + alfa-queroglutarato = oxaloacetato + glutamato
- é encontrada em altas concentrações no citoplasma e nas mitocôndrias do fígado, músculos esquelético e cardíaco, rins, pâncreas e eritrócitos (glóbulos vermelhos do sangue); quando qualquer um desses tecidos é danificado, a AST é liberada no sangue
- como não há um método laboratorial para saber qual a origem da AST encontrada no sangue, o diagnóstico da causa do seu aumento deve levar em consideração a possibilidade de lesão em qualquer um dos órgãos onde é encontrada
- valores normais: até 31 U/L (mulheres) e 37 U/L (homens)*
Estrutura molecular em 3D da aspartato aminotransferase
1.1.2 Alanina aminotransferase (ALT)
- também pode ser chamada de transaminase glutâmico pirúvica (TGP)
- é uma enzima que catalisa a reação: alanina + alfa-queroglutarato = piruvato + glutamato
- é encontrada em altas concentrações apenas no citoplasma do fígado, o que torna o seu aumento mais específico de lesão hepática; no entanto, pode estar aumentada em conjunto com a AST em miopatias (doenças musculares) severas
- valores normais: até 31 U/L (mulheres) e 41 U/L (homens)*
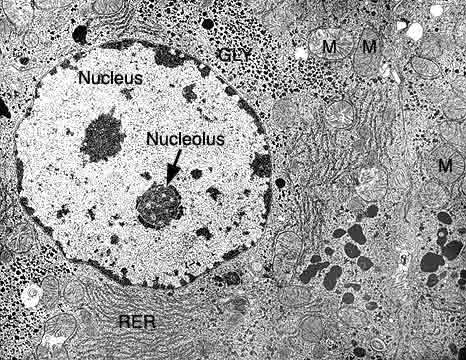
Microscopia eletrônica de hepatócito. As mitocôndrias (M) são organelas responsáveis principalmente pela produção de energia nas células. O citoplasma é o espaço da célula entre o núcleo (essa estrutura circular que ocupa grande parte da figura) e a membrana celular, que reveste a célula. É preenchido por uma matéria semi-fluida denominada hialoplasma aonde estão suspensas as organelas da célula.
1.1.3 Relação AST/ALT
- além das características individuais, a relação entre o aumento das enzimas tem valor diagnóstico
- tanto a AST quanto a ALT costumam subir e descer mais ou menos na mesma proporção em doenças hepáticas
- elevações pequenas de ambas, ou apenas de ALT em pequena proporção, são encontradas na hepatite crônica (especialmente hepatite C e esteato-hepatite não alcoólica)
- como na hepatite alcoólica há maior lesão mitocondrial, proporcionalmente, do que nas outras hepatopatias, observa-se tipicamente elevação mais acentuada (o dobro ou mais) de AST (que é encontrada nas mitocôndrias) do que de ALT, ambas geralmente abaixo de 300 U/L
- elevações de ambas acima de 1.000 U/L são observadas em hepatites agudas virais ou por drogas
|
Causas de aumento de aminotransferases no sangue |
|
| Doenças hepatobiliares | |
| Doenças do miocárdio | |
| Doença pancreática | |
| Doença muscular | |
| Álcool | |
| Ligação a imunoglobulina (rara) | |
| Doença não-hepatobiliar com envolvimento hepático | obesidade / diabetes |
| hemocromatose | |
| deficiência de alfa-1-antitripsina | |
| infecção pelo HIV | |
| hipertireoidismo | |
| doença celíaca | |
1.2 Desidrogenase lática (DHL)
- é observado em lesões hepatocelulares de modo geral
- pode ser útil na diferenciação entre hepatite aguda viral e lesão causada por isquemia ou paracetamol; sugere-se que, em elevações de aminotransferases acima de 5 vezes o limite superior, uma relação ALT/DHL maior que 1,5 sugere hepatite viral
- valores normais: 24-480 U/L*
- Testes para avaliação do fluxo biliar e lesão de vias biliares
2.1 Fosfatase alcalina
- trata-se não de uma enzima, mas de uma família de enzimas, presente em praticamente todos os tecidos; no fígado, é encontrado principalmente nos microvilos dos canalículos biliares e na superfície sinusoidal dos hepatócitos
- o aumento da fosfatase alcalina hepática é mais evidente na obstrução biliar, aonde o acúmulo de sais biliares a solubilizam e a obstrução promove a sua regurgitação entre as células hepáticas até o sangue
- em casos de elevação da fosfatase alcalina aonde não observa-se sinais clínicos ou laboratoriais de doença hepatobiliar, é possível a diferenciação entre as principais isoenzimas (hepática, óssea e intestinal) para localizar a fonte da alteração.
- valores normais variam de acordo com a idade: 1 dia de idade: até 250 U/L; 2 – 5 dias: até 231 U/L; 6 dias – 6 meses: até 449 U/L; 7 meses – 1 ano: até 462 U/L; 2 – 3 anos: até 281 U/L; 4 – 6 anos: até 269 U/L; 7 – 12 anos: até 300 U/L; 13 – 17 anos: até 187 U/L (mulheres) e 390 U/L (homens); adultos: 35 a 104 U/L (mulheres) e 40 a 129 U/L (homens)*
|
Causas de aumento “isolado” de fosfatase alcalina |
|
| Aumento da isoenzima hepática | Metástases hepáticas ou doença infiltrativa Cirrose biliar primária Colelitíase Aumento discreto com a idade |
| Aumento da isoenzima óssea | Fisiológica (infância, puberdade, pós-menopausa) Doença osteoblástica (Paget, osteomalacia, metástases) |
| Aumento da isoenzima intestinal | Doença hepática (cirrose) Diabetes mellitus Insuficiência renal crônica Doença intestinal ( linfoma, doença cadeia a ) Fisiológica ( discreta ) – aumento com ingesta de gorduras Secretores de sangue grupo O e B |
| Isoenzima placentária | Gestação normal Doença maligna (discreto) Cirrose infantil indiana |
| Formas variantes ou não usuais | Ligada a imunoglobulinas (doença autoimune, doença inflamatória intestinal) Derivada de tumores ( ovariano, testicular, hepatocarcinoma ) Fígado-símile mais osso (hiperfosfatasemia benigna transitória – aumento severo) |
| Geneticamente determinado | Qualquer das isoenzimas |
2.2 Gama glutamiltransferase (GGT)
- é uma enzima encontrada em grande quantidade no fígado, rins, pâncreas, intestino e próstata, mas também está presente em vários outros tecidos
- apesar de elevações muito grandes estarem associadas principalmente a câncer primário ou secundário do fígado e a obstrução biliar, alterações menores são poucos específicas de doenças do fígado; por outro lado, é um marcador muito sensível de doença hepática, pois está alterado em 90% dos portadores de doença hepatobiliar
- observa-se que cerca de 15% das pessoas tem a GGT acima dos valores considerados normais sem a presença de qualquer doença, mesmo com valores acima de 100 U/L
- elevações da GGT também podem estar associadas, sem nenhum significado patológico, ao uso de álcool e algumas medicações
- valores normais: 8 a 41 U/L (mulheres) e 12 a 73 U/L (homens)*
|
Causas de aumento plasmático de gama glutamiltransferase |
|
| Doença hepatobiliar | |
| Doença pancreática | |
| Álcool | |
| Drogas ( especialmente indutores enzimáticos, como barbitúricos) | |
| Doenças não hepatobiliares com envolvimento hepático (aumento leve) | Anorexia nervosa Distrofia miotônica Síndrome de Guillain-Barré Hipertireoidismo Síndrome metabólica Após infarto do miocárdio Porfiria cutânea tarda |
| Doença neurológica (aumento leve) | |
| Doença maligna / radioterapia | |
2.3 Bilirrubinas
- A bilirrubina, principal componente dos pigmentos biliares, é o produto final da destruição da porção “heme” da hemoglobina e outras hemoproteínas. A primeira bilirrubina a ser produzida nesse processo é a bilirrubina indireta (também chamada de bilirrubina não conjugada). Essa bilirrubina sofre o processo de conjugação e passa a ser bilirrubina direta (ou conjugada);
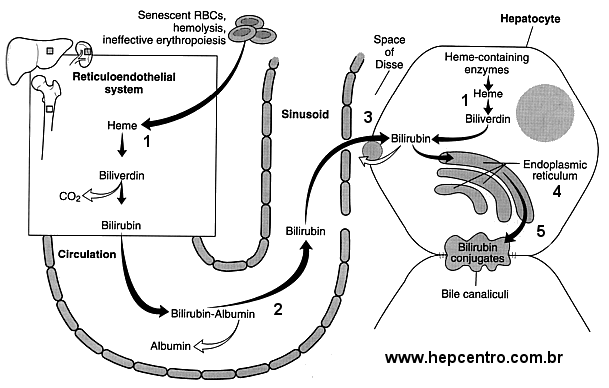
Metabolismo da bilirrubina: (1) destruição do heme e formação da bilirrubina; (2) transporte da bilirrubina pelo plasma, ligada à albumina; (3) captação da bilirrubina do plasma pelo hepatócito; (4) conversão no hepatócito da bilirrubina não conjugada em conjugada; (5) transporte da bilirrubina conjugada pela membrana biliar.
- os termos “direta” e “indireta” referem-se ao método criado para diferenciá-las por van den Bergh e Muller em 1916, mas que persistem até hoje (gerando confusão desnecessária);
- o aumento da bilirrubina indireta, portanto, é causado pelo aumento da degradação do heme ou deficiência da conjugação no fígado;
|
Causas de hiperbilirrubinemia |
| Não-conjugada (pré-microssomal) Produção excessiva de bilirrubina (hemólise) Hematopoese inefetiva Distúrbios hemolíticos Metabolismo anormal de bilirrubina (congênito) Imaturidade dos sistemas enzimáticos Icterícia fisiológica do recém nascido Icterícia da prematuridade Defeitos herdados Síndrome de Gilbert Síndrome de Crigler-Najjar Efeito de drogas |
| Conjugada e não conjugada (pós-microssomal) Distúrbio hepatocelular Doença hepatocítica primária (cirrose, hepatite, neoplasia, drogas) Colestase intra-hepática (drogas, colestase) Icterícia pós-operatória benigna Hiperbilirrubinemia conjugada congênita Síndrome de Dubin-Johnson Síndrome de Rotor Obstrução mecânica dos ductos biliares (icterícia obstrutiva) Extra-hepática (cálculos, neoplasia, estenose, atresia) Intra-hepática (colangiopatia obstrutiva infantil, colangite esclerosante, CBP) |
- o aumento da bilirrubina direta é causado principalmente por deficiência na eliminação da bilirrubina pela bile;
- o aumento de ambas pode ser causado por obstrução do fluxo de bile (mas com predomínio do aumento da bilirrubina direta) ou por lesão mais intensa dos hepatócitos (onde há deficiência na conjugação e também refluxo da bilirrubina conjugada para o sangue);
|
Causas de aumento da bilirrubina conforme a bilirrubina predominantemente aumentada |
|||||
| Bilirrubina não-conjugada (indireta) | Bilirrubina conjugada (direta) | ||||
| Aumento da produção de bilirrubina | Hemólise | Eritropatias | Doença do fígado | Doença hepatocelular (ex: hepatites) | |
| Hiperesplenismo, autoimune | Doença colestática (ex: CBP) | ||||
| Eritropoese ineficaz (ex: talassemias) | Distúrbio do metabolismo | S. de Dubin-Johnson | |||
| Destruição de hematomas | Síndrome de Rotor | ||||
| Redução da conjugação | Hiperbilirrubinemia neonatal | Colestase benigna | |||
| Jejum | Colestase da gravidez | ||||
| Síndrome de Gilbert | Doenças extra-hepáticas | Doença do trato biliar (ex: tumor) | |||
| Síndromes de Crigler-Najjar | Doença pancreática (ex: carcinoma) | ||||
- assim, a dosagem das bilirrubinas é um exame que pode avaliar ao mesmo tempo lesão hepatocelular, fluxo biliar e função de síntese do fígado;
- valores normais em adultos: total : 0,20 a 1,00 mg/dL; direta : 0,00 a 0,20 mg/dL; indireta: 0,20 a 0,80 mg/dL*
- valores normais da bilirrubina total em recém-nascido prematuro: 1 dia: 1,00 a 8,00 mg/dL; 2 dias: 6,00 a 12,00 mg/dL; 3 – 5 dias: 10,00 a 14,00 mg/dL*
- valores normais da bilirrubina total em recém-nascido a termo: 1 dia: 2,00 a 6,00 mg/dL; 2 dias: 6,00 a 10,00 mg/dL; 3 – 5 dias: 4,00 a 8,00 mg/dL*
- Testes para avaliação da função de síntese do fígado
3.1 Fatores da coagulação e atividade de protrombina
- o fígado tem papel central na hemostasia – sintetiza a maioria dos fatores e inibidores da coagulação, além de algumas proteínas do sistema fibrinolítico e elimina enzimas ativas dos sintemas de coagulação e fibrinólise; assim, doenças hepáticas severas costumam cursar com alterações na coagulação;
- a falta de fatores da coagulação podem ocorrer por perda da função dos hepatócitos, mas também por falta de “matéria prima” para a sua síntese – a síntese da maioria dos fatores de coagulação é dependente da vitamina K, que não é produzida no nosso organismo e precisa ser absorvida da dieta;
- como a absorção da vitamina K é dependente da presença de sais biliares e a cirrose diminui a sua produção (especialmente nas doenças colestáticas, como a cirrose biliar primária e a colangite esclerosante primária), espera-se no cirrótico algum grau de deficiência dessa vitamina, que pode ser suplementada por via parenteral (injeção);
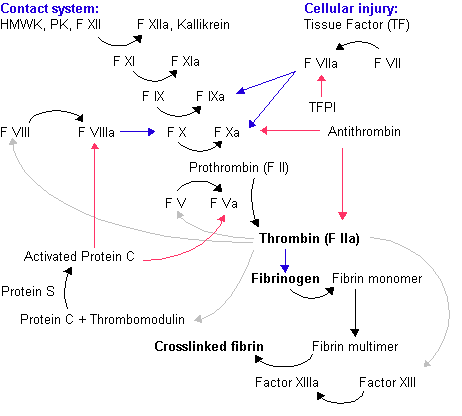
Cascata da coagulação. Note que a atividade da protrombina (ao centro) depende da presença de fatores da coagulação.
- na insuficiência hepática e/ou na deficiência de vitamina K, o primeiro fator a diminuir é o VII, seguido do II, X e IX;
- a síntese do fator V é independente da vitamina K; portanto, uma deficiência dos fatores II, VIII, IX e X sem deficiência do fator V (se essas deficiências se mantiverem após suplementação da vitamina K);
- na prática clínica, a determinação da atividade da protrombina (ou tempo de protrombina) é um método simples, barato e facilmente realizável para avaliar o conjunto dos fatores de coagulação e, portanto, da função de síntese do fígado;
- apesar da perda de fatores de coagulação e a redução da quantidade de plaquetas pela hipertensão portal e hiperesplenismo, essas deficiências são compensadas pelo aumento de fatores pró-coagulação, o que faz com que o cirrótico, mesmo com atividade de protrombina reduzida, tenha tendência a coagulação ao invés de sangramento – essa diferenciação é muito importante em algumas situações como cirurgias em portadores de cirrose, e pode ser avaliada pelos exames de tromboelastografia e tromboelastometriaç
- os valores normais de tempo de protrombina estão entre 11,1 e 13,2 segundos e são comparados em relação a plasma controle, analisando-se o tempo de atraso em relação ao controle ou através do RNI (international normalized ratio) que normalmente está entre 0,9 e 1,1*
3.2 Albumina
- a albumina é a principal proteína circulante no organismo humano e é responsável entre outras coisas, pelo transporte de substâncias (entre elas medicamentos) pelo sangue e pela maior parte da pressão coloidosmótica do plasma;
- o fígado é o único órgão responsável pela produção da albumina; reduções na quantidade da albumina no sangue (hipoalbuminemia), no entanto, podem não ser causadas por doenças do fígado, mas também por falta de “matéria prima” para a sua síntese (como nas desnutrições protéicas) ou aumento na sua destruição (estados catabólicos intensos) ou perda (intestinal ou renal);
|
Causas de hipoalbuminemia sérica |
| Diminuição de síntese Desnutrição Malabsorção Doença hepática Doença maligna |
| Aumento da perda Proteinúria (síndrome nefrótica) Enteropatia perdedora de proteínas – DII Queimaduras Doença exsudativa da pele |
| Aumento do catabolismo Estados hipercatabólicos (traumatismos, pós-cirurgico) |
| Erro da distribuição intra/extravascular (aumento da permeabilidade vascular) Estados inflamatórios (reação de fase aguda) |
| Hiperidratação |
| Variação genética Analbuminemia |
| Síntese interrompida Condições inflamatórias agudas e crônicas |
- como a meia-vida da albumina é relativamente alta (cerca de 20 dias), a redução da síntese pelo fígado pode demorar vários dias para se manifestar laboratorialmente (pela dosagem da albumina no sangue) ou clinicamente (especialmente pela formação de edema e ascite);
- na cirrose, excluindo-se outras causas, a hipoalbuminemia reflete principalmente a redução a síntese pelo fígado com alguma influência da desnutrição, que pode ser decorrente também da doença hepática; assim, a dosagem da albumina sérica tem importância dupla na avaliação do estágio da cirrose, participando do cálculo das classificações de Child-Pugh e do MELD.
- valores normais: 3,5 a 5,2 g/dL*
- Testes para avaliação de complicações e estágio da cirrose
4.1 Classificação de Child-Pugh
- a classificação de Child-Pugh é uma tentativa de agrupar em uma única classificação alguns dos fatores que seriam mais significativos no paciente com cirrose para prever o risco de submeter esses pacientes a um tratamento cirúrgico;
|
Classificação de Child-Pugh1 |
|||
| Encefalopatia hepática2 | ausente | 1-2 | 3-4 |
| Ascite | ausente | leve | moderada/severa |
| Albumina | > 3,5 | 3,0-3,5 | < 3,0 |
| Bilirrubina total3 | < 2,0 | 2,0-3,0 | > 3,0 |
| Tempo de protrombina4 | 1-4 | 4-6 | > 6 |
| Pontos: | 1 | 2 | 3 |
|
A: 5-6 pontos |
B: 7-9 pontos |
C: 10-15 pontos |
Notas: 1soma-se os pontos para cada um dos cinco itens; 2classificação de West Haven; 3na cirrose biliar primária, utilizar os seguintes valores de bilirrubina total: 1-4 (1 ponto), 4-10 (2 pontos) e > 10 (3 pontos); 4segundos após o controle – é possível também utilizar o valor de RNI: < 1,7 (1 ponto), 1,7-2,3 (2 pontos) e > 2,3 (3 pontos)
- a classificação, no entanto, tem sido utilizada por décadas na prática hepatológica como um modo de classificar, ainda que de modo grosseiro, o paciente cirrótico em três estágios (A, B e C), com grau progressivo de complicações da cirrose;
- mesmo assim, a classificação de Child-Pugh é incapaz de prever o prognóstico (expectativa de vida), com um mínimo de precisão, quando avaliada individualmente; a tendência atual é a de utilizar a classificação de MELD/PELD e abandonar a de Child-Pugh.
4.2 Alfa-fetoproteína
- é uma proteína que pode estar aumentada em 70-90% dos pacientes com carcinoma hepatocelular; apesar de ter valor limitado (também está aumentada na hepatite crônica em atividade e nem todos os hepatocarcinomas aumentam a proteína), a associação da dosagem de alfa-fetoproteína e exame de imagem (preferencialmente ultrassonografia) foi por muito tempo recomendada periodicamente em pacientes cirróticos para o diagnóstico precoce de câncer
- atualmente os protocolos de rastreamento de hepatocarcinoma não indicam a sua dosagem, bastando exame de imagem como a ultrassonografia, mas por diversos motivos práticos ainda continua sendo usada
4.3 Plaquetas
- a redução na quantidade de plaquetas no sangue (plaquetopenia) é comum em portadores de doenças hepáticas crônicas por cinco mecanismos principais: aumento do sequestro e destruição pelo baço aumentado (hiperesplenismo), redução na produção pela medula óssea, deficiência de ácido fólico, destruição por mecanismos imunológicos e por coagulação intravascular disseminada;
- a plaquetopenia ainda é de importância no paciente portador crônico de hepatite C com indicação de tratamento com interferon, pois durante o tratamento costuma haver exacerbação dos mecanismos imunológicos envolvidos na destruição das plaquetas, podendo levar a plaquetopenia severa e a hemorragias;
- como a quantidade de plaquetas reflete e é grosseiramente proporcional ao grau de hipertensão portal (que leva à esplenomegalia e ao sequestro de certo modo proporcional ao aumento do baço), que por sua vez também é proporcional ao grau de fibrose hepática, a dosagem de plaquetas têm sido utilizada como método indireto de avaliação do grau de fibrose hepática e como preditor do risco de surgimento de varizes gastroesofágicas.
4.4 FibroTest®
- é um método não invasivo, criado por Imbert-Bismuth e colegas onde, através de um algoritmo matemático com base em cinco variáveis (bilirrubina total, GGT, haptoglobina, alfa-2-macroglobulina e apoliproteina A1), com resultado entre 0 e 1, procura-se estimar o grau de fibrose hepática;
- o método é muito preciso para o diagnóstico de ausência (com resultado < 0,1) ou presença (>0,6) de fibrose significativa, mas é pouco útil na avaliação de estágios intermediários;
- tem sido usado como opção à biópsia hepática pré e pós tratamento em pacientes com hepatites virais (especialmente hepatite C) e na avaliação de portadores de esteato-hepatite não alcoólica.
4.5 APRI
- o índice da relação aspartato aminotransferase sobre plaquetas (APRI) é um marcador de fibrose hepática em pacientes portadores de hepatite C;
- ao contrário do FibroTest, é baseado em exames comuns e baratos e seu cálculo não é patenteado, o que permite que seu cálculo seja gratuito e disponível em diversas calculadoras online ou em aplicativos;

- o APRI < 0,5 praticamente descarta fibrose significativa, enquanto > 1,5 praticamente confirma fibrose avançada;
- para fins de adquirir tratamento da hepatite C pelo SUS, por exemplo, o APRI é aceito como comprovação de fibrose avançada, sendo muito mais barato, não invasivo e acessível que as alternativas
4.5 MELD/PELD
- o Modelo para Doença Hepática Terminal (Model for End-Stage Liver Disease) é uma escala numérica criada para avaliação da gravidade da doença heática, em uma escala de 6 a 40, utilizando um algoritmo matemático baseado em três variáveis: bilirrubina total, RNI e creatinina (que mede a função do rim);
- o PELD (Pediatric End-Stage Liver Disease) é uma escala semelhante, criada para crianças com menos de 1 ano de idade, baseada em cinco variáveis: bilirrubina total, RNI, albumina, se há distúrbio de crescimento e se a criança tem mais ou menos que 1 ano de idade;
|
Algoritmo de cálculo MELD/PELD |
|
| MELD = 0,957 x Log e (creatinina mg/dl) + 0,378 x Log e (bilirrubina mg/dl) + 1,120 x Log e (INR) + 0,643 x 10 e arredondar para valor inteiro – Caso os valores de laboratório sejam menores que 1, arredondar para 1,0. – A creatinina poderá ter valor máximo de 4,0, caso seja maior que 4,0 considerar 4,0. Caso a resposta seja sim para a questão da diálise (realiza diálise mais de duas vezes por semana?), o valor da creatinina automaticamente se torna 4,0. |
PELD = 0,480 x Log e (bilirrubina mg/dl) + 1,857 x Log e (INR) – 0,687 x Log e (albumina mg/dl) + 0,436 se o paciente tiver até 24 meses de vida + 0,667 se o paciente tiver déficit de crescimento menor 2 x 10 – Caso os valores de laboratório sejam menores que 1, arredondar para 1,0. – Cálculo do valor do déficit de crescimento baseado no gênero, peso e altura. |
- o MELD e o PELD, pelo seu perfil de reprodutibilidade e disponibilidade, ganhou mais importância no Brasil em 29 de março de 2006, quando o Ministério da Saúde publicou a Portaria 1.160, modificando o funcionamento da ordem da lista de transplante de fígado de cronológica para gravidade, baseada neste critério;
- os cálculos do MELD e do PELD podem ser feitos online aqui.
BIBLIOGRAFIA
- Questions and answers for patients and families about MELD and PELD. Unos.org
- Fox RK, Wright TL, Viral Hepatitis in Current Diagnosis & Treatment in Gastroenterology
- Rosalki, SB e McIntyre, N em Bircher, J, Benhamou, JP et al. Oxford Textbook of Clinical Hepatology, Oxford Medical Publications,1999.
- Denninger, MH em Bircher, J, Benhamou, JP et al. Oxford Textbook of Clinical Hepatology, Oxford Medical Publications,1999
- Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg 1973; 60: 646-9.
- Thomopoulos KC; Labropoulou-Karatza C; Mimidis KP; Katsakoulis EC; Iconomou G; Nikolopoulou VN Non-invasive predictors of the presence of large oesophageal varices in patients with cirrhosis.Dig Liver Dis. 2003; 35(7):473-8
- Bressler B; Pinto R; El-Ashry D; Heathcote EJ Which patients with primary biliary cirrhosis or primary sclerosing cholangitis should undergo endoscopic screening for oesophageal varices detection? Gut. 2005; 54(3):407-10
- Laurent Castera, MD; Jean-Michel Pawlotsky, MD Noninvasive Diagnosis of Liver Fibrosis in Patients With Chronic Hepatitis C Medscape General Medicine
- Imbert-Bismut F, Ratziu V, Pieroni L, Charlotte F, Benhamou Y, Poynard T. Biochemical markers of liver fibrosis in patients with hepatitis C virus infection: a prospective study. Lancet. 2001; 357: 1069-1075
- Shah, N. L. and Caldwell, S. H. (2016), Assessing the risk of bleeding and clotting in cirrhosis. Clinical Liver Disease, 7: 26-28. doi:10.1002/cld.528
- Muciño-Bermejo J1, Carrillo-Esper R, Uribe M, Méndez-Sánchez N. Coagulation abnormalities in the cirrhotic patient. Ann Hepatol. 2013 Sep-Oct;12(5):713-24. (link)
- Clinical Practice Guidelines Management of Hepatocellular Carcoinoma – EASL (link)
OBSERVAÇÕES
* Os intervalos considerados como referência nesse artigo são os utilizados pelo Laboratório Fleury apenas para fins didáticos. Esse intervalo pode variar de acordo com o método e aparelhagem utilizados, portanto é mais confiável se basear nas referências do laboratório onde o exame foi realizado. Não há nenhuma relação comercial entre o Hepcentro e o Laboratório Fleury.


